
How does a GC-MS machine know that there’s a drug in the blood?
The analyst has to teach it. Just like you teach a kid his or her colors, you have to teach this machine what are the drugs it should be looking for. How were you taught what color is what? You probably don’t remember yourself it was so long ago, but if you have any children you likely remember that it was a process. Perhaps you used flashcards or marbles or blocks, but the basic process is to show the toddler a standard (“this is red”) with the stated goal that every time the child sees something just like that standard, they report what they see as “red.” Then, you move on to another color. You show the toddler another color (e.g., yellow) and tell the toddler what that different hue is (e.g., yellow). If properly taught and if it “sticks” in the toddler’s mind, the toddler has learned what is red and what is yellow, but most importantly the toddler is taught that what is red is not yellow. These are mutually exclusive thoughts when learned. That which is red cannot also be yellow and vice versa. As the toddler becomes more proficient at learning his or her colors, you introduce shades of the primary color. For example, you would show red, then show and teach the toddler pink or burgundy or cardinal and the like. Things that are close to red, but are not red. This way the toddler is becoming specific to your teaching of what red is. He or she when shown pink will not reply red because pink and red are now different.
If you mistakenly teach the toddler that yellow is blue. He or she will report that yellow is blue through no fault of his or her own, but because you taught the toddler wrong. The toddler will not be lying; the toddler will not be trying to trick or deceive. Even when pressed will believe with his or her whole heart that it is the wrong color. This process of teaching is very important.

Arriving at the proper qualitative result in Driving Under the Influence of Drug (DUID) cases is a process. If each part of the process does not work correctly, then there is no validity in the call that there is a drug present in the blood or urine of the accused. Urine should never ever be used to try to determine whether or not a motorist is impaired. For impairment, urine tests whether they are for drugs or ethanol are all but worthless.
Step 1: Presumptive Screening
In a typical laboratory, the first analytical step in determining whether or not drugs are present in the blood or urine is to do enzyme screening (EMIT, CEDIA or ELISA based). This is a colormetric (change of color) based test. A very small portion of the blood is sampled. These simple and quick color tests are very basic. They are prone to false positives and false negatives.
What the laboratory analyst is doing in this step is exercising judgment. In a colormetric testing, a blank is prepared (one where there are no drugs at all that react), and tested. It must come up with a negative result. A standard (a known drug) is prepared and tested. The standard must react and change color to the targeted color that indicates a positive. The unknown (from the motorist) is compared to the blank and the standard. The truth is if the motorist’s sample is “close enough” to the standard, then the analyst will call this a “positive.” This subjective process is never memorialized in a meaningful way. There are no photographs or videos taken of the analysis or its results. It begs the question, other than the analyst’s say so, how do we know that this part of the analysis was ever preformed?
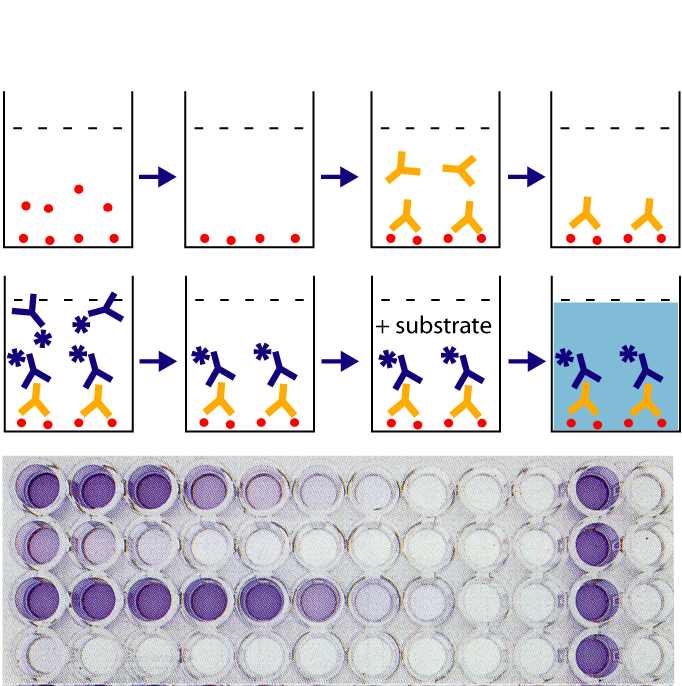
As you can see from the above photograph in Figure 1, these tests produce visible results. These results are never preserved in any way that is verifiable. They should be, but are not. The purpose for using this type of test is simply to help screen how the rest of our analysis should go. Should there be no more time invested because the presumptive test comes back negative? Or should more testing be conducted because there is a presumptive positive?
For those of you who really want to understand the enzymatic process, I offer my favorite video on the subject:
Video 1: How the enzymatic drug screening for DUID cases works
No matter what the context, a presumptive positive must never be used as proof presence of a drug in the blood or urine. Too frequently in work place testing, probation and parole violations and even DUID contexts, laboratories use this inferior form of testing alone and report presence. People should never be punished based upon these results alone. The very most that these results will give us is a “presumptive positive” for a class of drugs. There is a lot of literature, and many blog posts on this site, that discusses known false positives. This is why additional testing must always be carried out. Colormetric drug screening tests can and never will be able to tell us for certain that that drug is there. We need to do more testing by a more specific and more objective means.
Step 2: GC-MS
When there is a motorist accused of the charge of DUID, the primary means of confirmatory testing and identification of whether or not there is a drug present in the blood or urine, and if so, by how much, is the Gas Chromatograph with Mass Spectrometer (GC-MS) device. To achieve a proper result using a GC-MS, there is an involved process.
At its basic core, the concept of teaching the machine is not unlike teaching a child its colors.If you teach the machine wrong or if you only partially test the machine or rely on only part of its process, there will be no validity in the results. There is a myth in the courtroom that the GC-MS machine is the “gold standard” in forensic science and that it simply produces binary concepts that require no interpretation like a calculator when asked to process 1+1. Nothing can be further from the truth. It is a subjective opinion that the analyst is making at the end of the process. It is a “best guess” based upon empirical data. But like all matters of interpretation, it can be clouded or confused by forms of cognitive bias as well as lack of proficiency or understanding. While there is generated data, at the end of the analysis, this process is far from simply a mathematical process. It is interpretive.
This second step has different sub-steps to it. All of the sub-sets must be present in order for there to be a valid call as to presence of a drug, and then if validly detected, how much there is. Each sub-part cannot correctly identify and quantify a drug alone. All of the sub parts must be correct. Think of it like a Venn Diagram. It is only at the convergence all all of the data, that we can take the GC-MS data and validly opine with some degree of confidence that a given drug in an unknown is or is not present and by how much.
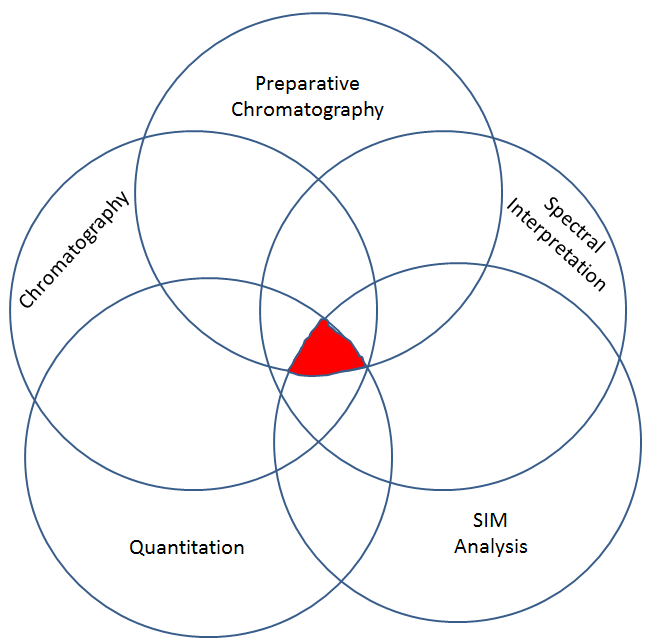
Step 2, part 1: Preparative Chromatography
First the analyst has to extract (remove), the drugs from the matrix (the blood). The analyst cannot simply inject the blood or urine directly into the GC-MS. There is a great deal of preparative chromatography that needs to be completed with the blood or urine sample before it can be injected into the GC-MS.
The preparative chromatography process involves many steps: the primary steps are called extraction and derivatization.
In an extraction, the analyst uses chemicals to form the sample into layers. The making of layers is an important step in the process. The unionized drug distributes into the organic phase (likes dissolves likes) due to it more lipophilic nature. When the drug was ionized it was more hydrophilic since it was soluble in the water. By making the aqueous solution basic a base drug is put into its unionized form and if the solution is made acid any acidic drug is placed into the unionized state. The organic layer is removed and evaporated to dryness under nitrogen and then the derivatization is done.
Some drugs of abuse are not volatile organic compounds (VOCs). Gas Chromatography only works on VOCs. So, we have to change the drug to detect the drug if we want to use GC. This changing of the drug to make it into a VOC so that the GC will work is called derivatization. We have to “tie up” the OH groups (hydroxyl groups), COOH groups (carboxylic acid groups), and the like. Yes, we in essence totally change the molecule so we can detect it. It is the reproducibility of running standards that gives the confidence that although we are changing the drug, this change is consistent and reproducible. If we run our blanks, controls (calibrators, verifiers), and unknown samples through this same process of derivatization that has been proven in the past by a validation study to be stable, consistent, and reproducible, then we can have confidence in the result.
The other reason to use derivatization is to provide for better chromatography–to try to get tall, skinny, symmetrical peaks with good separation (resolution). It reduces the tailing of polar compounds of drugs that contain OH, –COOH, =NH, –NH 2 , –SH, and other functional groups.
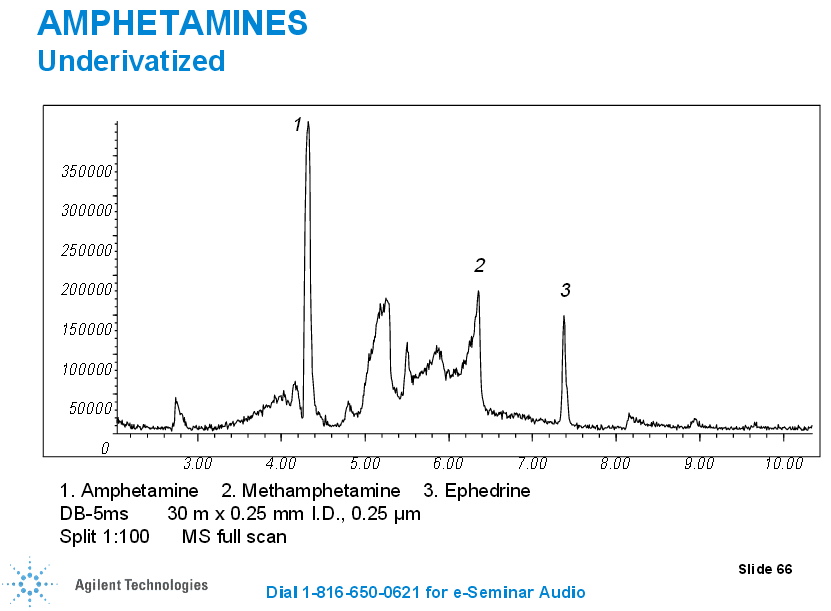
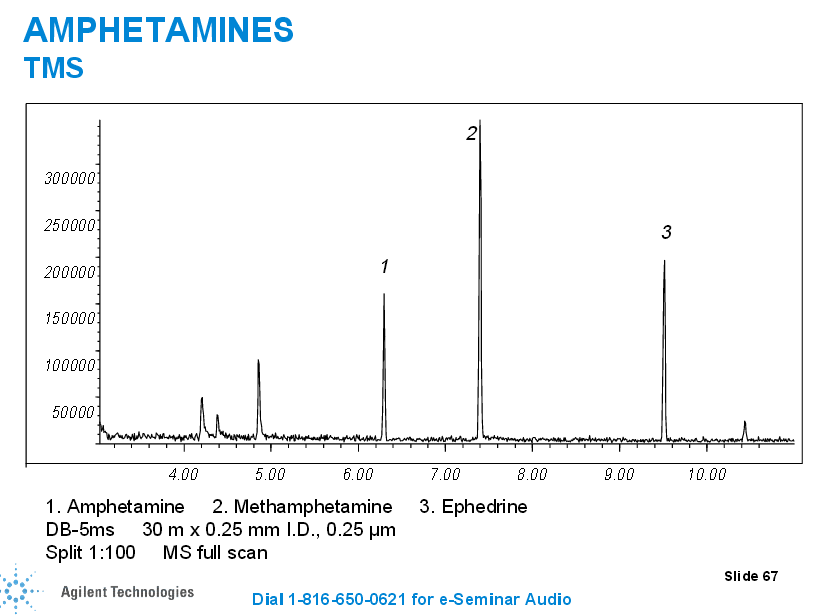
In the case of some drugs, such as the case with opioids, we may have to complete a double derivatization process (the second derivatization is for propionyl groups) so we can be highly selective for one specific opioid (we can distinguish between different opiates such as hydrocodone, hydromorphone, oxycodone, noroxycodone, and oxymorphone and those that may interfere without the second derivatization morphine and codeine).
In the case of opioid detection, this derivatization process can include steps such as:
- an extraction from the biological matrix (i.e., blood or urine) following deprotenization (for example using acetonitrile and centrifuging) (making supernate, not whole blood),
- oxime derivatization step (for example, using acetate buffer with hydoxylamine) by Solid Phase Extraction (SPE).
- The resultant is then derivatized a second time (for example TMS Derivatization using acetonitrile, BSTFA and TCMS), and
- analyzed by instrumental means using the GC-MS.
Step 2, part 2: Chromatographic Separation
If you can’t pass GC, you have no GC-MS—Heather Harris, MFS.
The very important first step in the GC-MS process is chromatographic separation. This is just simply good old fashion adsorption based chemistry based upon the stationary phase of the wall coated open tubular (capillary) column that is selected by the analyst. The selection of the column is very important. Different columns separate differently based upon the stationary phase. This is where the results from the drug screening by colormetric (enzymatic) means comes into play. If we have a presumptive positive result, we can be more selective on the column used.
This first instrumental step results that there is a determination based upon retention time or relative retention time. The retention time is then compared against a standard from a known from a trusted source. This is the traditional means of non-specific identification that is used in a blood ethanol analysis. A retention time identification is only characteristic of a compound based upon the same retention time of the standard. Retention time never ever proves identification by itself.
It is important to note, that the retention time MUST match (or be within the laboratory’s arbitrary acceptance criteria, making the retention time really a retention window). If the retention time for the unknown and the standard do not match, it cannot be that drug. Stated differently, it does not matter if the other steps that come afterwards come up with a near perfect match based upon diagnostic ions or even the full spectral scan, the retention time must match. You can look to see if the retention time matches by looking for the two Total (reconstructed) Ion Current Chromatograms (TICs). There must be one for the standard. There must be one for the unknown. You simply overlay them. If they do not match or are outside of the retention time window, then the laboratory cannot make the call that it is that drug.
For example, let’s consider this TIC that has standards involving drugs of abuse:
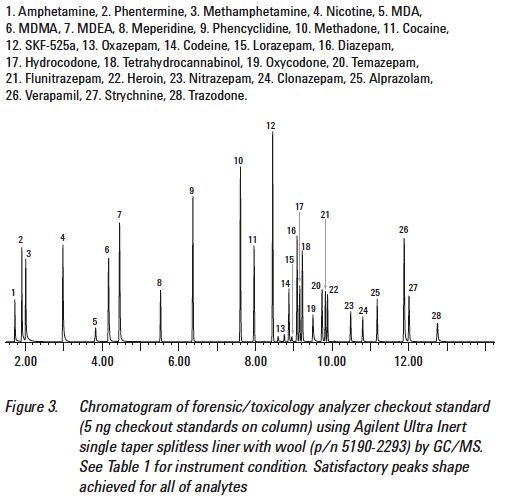
Consider when you get your client’s TIC in comparison.
As we can see from the above Figure 5, the peak associated with the amphetamine standard occurs at less than 2 minutes. When we look at Figure 6, the motorist’s TIC, there is a very large peak at 7 minutes. If the laboratory claims that there was amphetamine present in the sample that gives rise to the TIC seen in Figure 6, the laboratory is utterly wrong. It cannot be amphetamine because the peak that is associated with the standard for amphetamine does not match the peak that is with the unknown.
Retention times much match.
Step 2, part 3: Spectra Interpretation
The TIC is then developed into a spectra at a retention time using the scan function. This is a simple hunt and peck process by the analyst. The analyst simply right double clicks on his or her mouse at a given point on the spectrum, regardless of the retention time matching, and then processes it through the installed libraries.
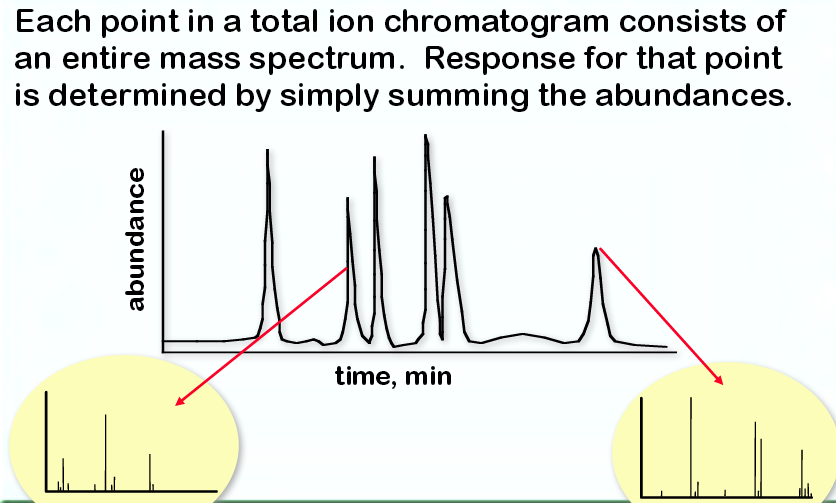
In essence, this step is a computerized search from a database. It takes previously loaded standards and compares them against your unknown. Here is an example of developed GC-MS spectrum when the machine is set to scan from a TIC:
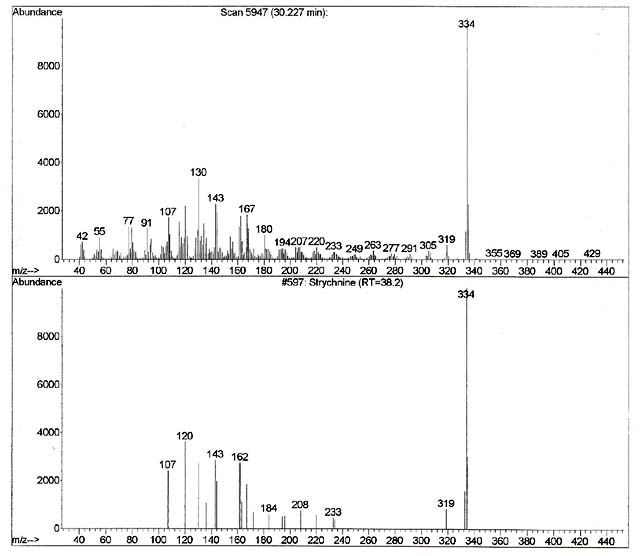
As discussed above, our first step in confirmation of the computer result is to see if the retention time of the TIC matches. As one can see on the top spectrum in figure 8, the spectrum associated with the unknown was generated at retention time 30.227 minutes. Also, one can note that the bottom spectrum comes from a local library that has been developed on this machine using this method. The retention time generated with the strychnine standard that was previously run using this particular instrument and this particular method reveals a retention time of 38.2 minutes. The retention times do not match. It is even outside of the published guidelines for the laboratory in terms of its retention time window, which was +/-0.5 minutes. Therefore, even if the two spectra identically matched, which they clearly do not in this case, the unknown in the blood cannot possibly be strychnine as the retention times do not match. Stated differently, no matter how beautiful of a match the spectrum from the unknown is when compared to the standard, if they do not match in terms of their retention times (retention time windows), the detected drug that is the library’s best match cannot be that drug. The retention times much match.
If this first step of confirmation of the retention time is not met, our theory that it is strychnine has been falsified. It cannot be strychnine.
If we do have a retention time (retention time windows) match, then we must next evaluate the unknown spectrum to the spectrum from the known. The computer usually does this, but a human being must also examine it to make sure the result “makes sense.” The fragmentation pattern must be explainable by the analyst. For our strychnine example, the analyst must be prepared to explain why the chemical with the formula of C21H22N2O2 and the molecular weight of 344 fragments into those discrete ions that are in the standard. They must be able to explain logical loss patterns from that molecular ion. If the analyst cannot explain how the standard spectra “makes sense” in terms of the fragmentation pattern through logical loss, rearrangements, or isotopic issues, then that is not acceptable.
As we can see from the two spectra in Figure 8, the computer keyed in on the 344 ion which is the molecular weight of strychnine. A lot of the other fragments that are present in the spectrum for the standard (208, 184, 162) are missing all together in our unknown. The analyst needs to explain why they are missing. This explanation has to be scientific and with supportable sources other than “trust me” as to why those ions are missing. Further, as we can see from the unknown spectrum there is a whole bunch of other ions detected that are not in the standard. Again, the analyst needs to explain that scientifically and with supportable sources other than “trust me” as to why those ions are there. Specifically troubling are the ions beyond the molecular ion of 344. The analyst needs to explain that scientifically and with supportable sources other than “trust me” as to why those ions are there when they “weigh more” than the chemical does in nature. It’s not going to be all column bleed (erosion of the GC-MS column) as that is most frequently at 44, 207, 221, 281, and 295 or through derivatization. Perhaps the 355 and 429 are from column bleed, but not the others. So an explanation that is scientifically defensible and supportable by external references is due.
One of the tip offs for me in this case is that there is an issue in addition to the incorrect retention time is that there was no reporting of the computer algorithm used to compare the unknown and the standard. Typically, there will be a report of the “goodness” of the computer search. The most popular ways of computer searching are using the NIST search or the PBM search. The result is called a match factor or quality score. With the advent of synthetic drugs, such as synthetic cannabinoids and synthetic cathinones, there is an excellent argument that the match factor and quality score are not the best means to identify and unknown as the use of the match factor or quality score presumes that the drug that is in the unknown has been identified and is in your library on your machine. It could be argued that the more realistic measurement of degree of “match” is the probability score as the probability score does not presume that the drug that is detected is in your library.
There are many frequent mistakes at this stage:
- The laboratory just skips over this step. They do not do an analysis in scan mode, and instead simply skip to Step 2, part 4: SIM analysis.
- The laboratory relies on an outdated spectral library to do the search.
- The laboratory does not conduct its own fragmentation experiments using its own instruments using certified reference materials to make its own library that is unique to its validated method and MS.
- The laboratory edits the report to not give information of the source of the standard it is comparing against the unknown, or fails to provide the hit list (the possible top 100 substances identified) in its data.
- The laboratory only reports the quality score or match factor, and not the probability score.
- The analyst gets an acceptably high match factor, but does not check the retention time (remember, the retention time must match).
Step 2, part 4: SIM analysis
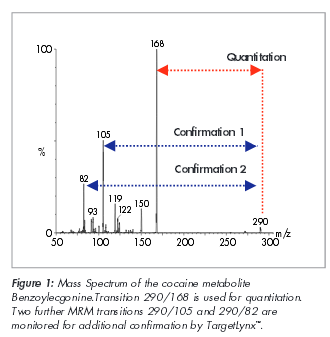
SIM is an acronym that stands for Selective Ion Monitoring. Unlike the scan setting that is described in step 2, part 3 supra, the GC-MS is programmed to ignore all fragments except for those that are pre-selected before the analysis to detect (monitor). With SIM analysis, you are losing a whole of of information as we can see below.

The analyst simply presumes that the 2 or 3 or 4 targeted and detected ions are unique and diagnostic to a particular drug to the exclusion of all others in the univers. I have pressed and pressed and pressed people for any sort of validation that proves this presumption. To date, no one has provided it to me.
Some crime laboratories, not all, add the quality assurance step that requires that the targeted ions not only be present, but that they are within certain ratios of one another. They take the SIM, they change the y-axis to relative abundance, then they compare (using a computer typically) the abundances to one another to see if they are within certain ratios. They likewise presume that if the diagnostic (target) ions are present and within certain ratios, then it is definitely the drug and nothing else. I have pressed and pressed and pressed people for any sort of validation that proves this presumption. To date, no one has provided it to me.
One of the problems that are typically seen in forensic laboratories is that the targeted ion may be there on the SIM, but the ion is below the accepted signal-to-noise ratio for that particular instrument running that particular method. So when analyzing SIM data, one needs to know what the raw (non-relative abundance) signal is for the SIM and also the signal to noise ratio.
As in all good science, until I can be provided with a robust validated and well-researched study that proves otherwise, the unvarnished scientific truth is that the presumption that the 2, 3 or 4 diagnostic ions that are targeted even with the ratios being as expected are NOT specific to only one drug to the exclusion of all other drugs. Please show me how I am wrong based on science.
One of the benefits that I have in having a national practice is that I get to see a lot of methods. By far, the most popular drug that is in DUID cases is Δ9-TCH or one of its metabolites. Different laboratories have different target ions for this same drug, and if they even bother to do ion ratio analysis, they also have different ratios for the same drug from laboratory to laboratory. So, is this the case that the emperor has no clothes? Perhaps. I hope not.
The saving grace may be the orthogonal approach where the Venn Diagram works or more particularly the consistent results that come from the retention time matching, the scan spectrum analysis, plus the SIM all meeting criteria.
Step 2, part 5: Quantitation
The final step in the analysis is the quantitation of the drug detected. Remember, if you do not have evidence and proof of retention time matching, the scan spectrum analysis, plus the SIM all meeting criteria, then you cannot and must not quantitate. The process of quantitation is just like our familiar method used in GC-FID for ethanol. We have to take different samples at different concentrations using certified reference materials (CRMs) and introduce them into the machine. We have to cover the linear dynamic range of expected results. We have to evaluate the linearity by using acceptable statistical methods (e.g., r2>0.999). We also use internal standards. Sometimes we use deuterated internal standards. The best internal standard for DUID analysis is an isotopically labelled version of the molecule you want to quantify. Ideally in DUID analysis, a deuterated internal standard will have the same extraction recovery, ionization response, and the same chromatographic retention time. Unlike in GC-FID, you want the deuterated internal standard to co-elute with the compound to be quantified. Because we use SIM analysis, we will be able to separate out the deuterated internal standard from our targeted drug that elutes from the column at the same retention time because the deuterated internal standard will have different mass charges (an increase by 3 to 5 mass charge units because deuterium has a higher mass than hydrogen). The GC-MS will be able to separate the deuterated internal standard from the unknown that is in the blood or urine.
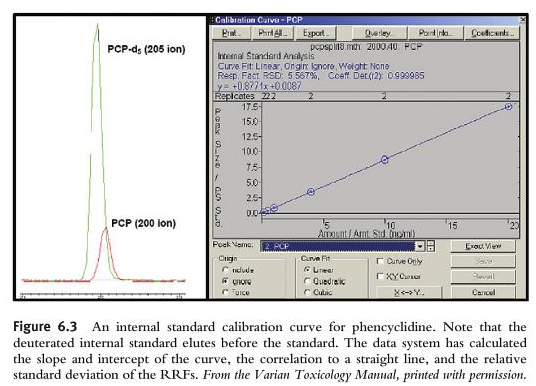
As we learned about SIM analysis above, the entire information of the mass spectrum when set to scan is missing in favor of simply 2, 3 or 4 claimed diagnostic (target) ions. However, the laboratory does not quantify on all of the targeted ions. It only chooses one. The quant ion is monitored in SIM, it is then used to produce a SIM chromatographic peak.
So, to verify the result, we need to inspect the calibration curve that is generated for its linearity. We must also inspect the quantitation report for errors (e.g., was it at the right retention time, were all of the target ions present, were they in the right ratio).
Remember that in order to quantitate it MUST first have data that proves that retention time matching, the scan spectrum analysis, plus the SIM all meeting criteria (diagnostic ions and ratios).
Conclusion
This is a very brief overview of how DUID testing is performed in the United States. There is a lot more to it. For the sake of justice, we who are involved in the criminal justice system cannot simply trust the laboratory. We can trust, but we must verify. Justice demands no less.

 The McShane Firm | DUI Lawyers
The McShane Firm | DUI Lawyers Pennsylvania DUI Blog
Pennsylvania DUI Blog
JR says:
Mr. McShane,
You made it understandable. Great explanation! Thank you!
Justin J. McShane says:
Thank you for the kind words. I saw that there was no place on the internet that explained it. So, I built it. I am glad that you found some value in it.
Lily hemlesly says:
Lol I <3 this website, it's gr8;)